Über Cystinose
Cystinose im Überblick
Was ist Cystinose?
Cystinose ist eine lysosomale Speicherkrankheit und gehört zu den Seltenen Erkrankungen.
Die Erkrankung ist vererbbar und verläuft progredient, d.h. es handelt sich um eine voranschreitende chronische Erkrankung. Die Schätzungen, wie viele Menschen weltweit von Cystinose betroffen sind, liegen zwischen einer Person von 100.000 und einer Person von 200.000.1 Cystinose kann den ganzen Körper betreffen und sich so auf unterschiedliche Organe und Gewebe auswirken. Die ersten Symptome betreffen häufig die Nieren und treten meistens bereits im frühen Kindesalter auf.1
Ursachen für Cystinose
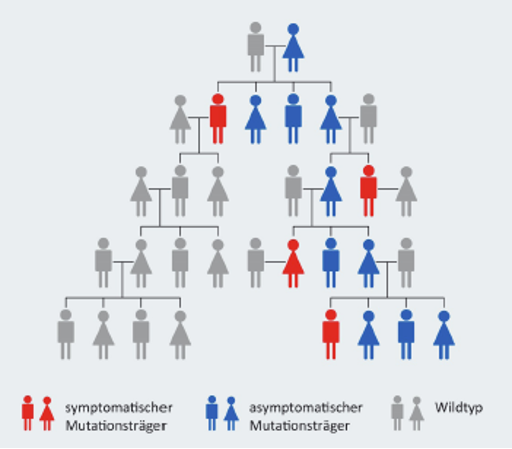
Der Auslöser für Cystinose sind Mutationen, also Veränderungen, in einem bestimmten Abschnitt des Erbmaterials. Deshalb spricht man von einer vererbbaren Erkrankung. Sie tritt aber nur auf, wenn die Mutation von beiden Elternteilen weitergegeben wird.
Mediziner*innen sprechen in einem solchen Fall von einem autosomal-rezessiven Erbgang.
Was passiert bei Cystinose im Körper?
Die Erkrankung nimmt ihren Anfang in den Lysosomen, den „Recyclinghöfen“ der Zelle. Hier werden körpereigene und körperfremde Materialien abgebaut und zu einzelnen Bestandteilen recycelt. Die kann der Körper dann entweder wiederverwenden oder ausscheiden. Einer dieser Bestandteile wird Cystin genannt. Normalerweise wird das Cystin von einem Transporter, dem Cystinosin, aus den Lysosomen heraustransportiert. Durch die Genmutation ist dieser Transporter jedoch defekt. Dadurch verbleibt das Cystin in den Lysosomen und bildet Cystinkristalle. Diese Kristalle wiederum schädigen die Zellen und damit schließlich unterschiedliche Organe wie z.B. Nieren oder Augen.
Wenn Sie mehr über Symptome und Auswirkungen erfahren möchten, klicken Sie hier.
Cystinose ist nicht gleich Cystinose
Wie Cystinose in Erscheinung tritt und wie sie verläuft, ist sehr individuell und hängt unter anderem von der Form der Erkrankung ab. Insgesamt gibt es drei verschiedene Formen von Cystinose, wobei 95 % der Betroffenen an der sogenannten nephropathischen Cystinose leiden.1 Deshalb finden Sie auf dieser Website vor allem Informationen zu dieser Form der Erkrankung.
Verschiedene Formen der Cystinose
Infantile Form (Nephropathische Cystinose)
Wie bereits erwähnt, ist sie mit etwa 95 % der Fälle die häufigste Form. Sie wird oft in den ersten zwei Lebensjahren diagnostiziert, meistens wegen bereits früh auftretender Nierenschädigungen. Um dem Auftreten eines Nierenversagens entgegenzuwirken, ist es wichtig, die nephropatische Cystinose frühzeitig zu behandeln.
Juvenile Form
In wenigen Fällen erfolgt die Diagnose erst später – meist im Jugendalter. Die Symptome sind mit denen der infantilen Form vergleichbar, der Verlauf der Erkrankung ist jedoch in der Regel milder, weil sich hier weniger Cystin ablagert.
Adulte Form (okulär, nicht-nephropathisch)
Diese Form der Cystinose ist die seltenste. Das Cystin wird hier erst bei fortgeschrittenem Alter des*r Betroffenen nicht mehr aus den Zellen abtransportiert. Somit wird die Diagnose erst im Erwachsenenalter gestellt, da sie sich lediglich in einer Ansammlung von Cystin-Kristallen in der Hornhaut äußert. Die Niere und andere Organe sind meist nicht betroffen.
- Nesterova & Gahl; Pediatr Nephrol. 2013;28(1):512–9.
- Gahl et al. N Engl J Med. 2002;347(2):111–121.
Symptome
Cystinose hat viele Gesichter
Auf den ersten Blick sind Cystinosepatient*innen meist kleiner und zierlicher als für ihr Alter üblich und besonders Patient*innen europäischer Abstammung haben oft eine blasse Haut, blaue Augen und blonde Haare. Bei der Cystinose kommt es zur Schädigung verschiedener Organe und Gewebe, weil sich in den Lysosomen der Zellen sogenannte Cystinkristalle bilden. Wenn Sie mehr dazu erfahren möchten, was bei Cystinose im Körper passiert, klicken Sie hier. Besonders früh und stark sind die Nieren betroffen, aber auch die Knochen und Augen werden stark in Mitleidenschaft gezogen. Aufgrund der Nierenschädigung kommt es zudem meist zu einer Wachstumsstörung.
Wie die Erkrankung verläuft und welche Symptome auftreten, ist je nach Form der Cystinose unterschiedlich.
Infantile Form (nephropathische Cystinose)
Mit 95 % der Fälle tritt die nephropathische Cystinose am häufigsten auf.1 Bei dieser Form sind zunächst besonders die Nieren betroffen, wo es zu schweren Funktionsstörungen kommt. Während die Lysosomen die Recyclinghöfe der Zellen sind, sind die Nieren die Filteranlagen des Körpers. Die Patient*innen leiden am Fanconi-Syndrom, das eine komplexe Funktionsstörung sozusagen direkt am Anfang der Filteranlagen darstellt und deswegen besonders schwerwiegend ist. Die Patient*innen scheiden vermehrt Wasser und Elektrolyte aus. Dadurch werden ihre körperliche Entwicklung und ihr Wachstum gestört. Das Fanconi-Syndrom wird im Alltag dadurch auffällig, dass die Patient*innen sehr häufig zur Toilette müssen, um Wasser zu lassen und sie deshalb auch immer viel Durst haben. Da dies zu einer Dehydrierung führen kann, ist es wichtig, dass Betroffene ausreichend trinken – sechs Liter Wasser pro Tag können bei Cystinose normal sein.
Die nephropathische Cystinose ist allerdings keine reine Nierenerkrankung. Gerade im weiteren Verlauf können auch ganz andere Symptome auftreten wie Muskelschwäche, die auch zu Atemproblemen und Schluckstörungen führen kann oder einer Schilddrüsenunterfunktion.
Insgesamt sind Betroffene schneller erschöpft und körperlich weniger belastbar. Eine typische Cystinose-Erscheinung sind auch Augenbeschwerden, wie z. B. Lichtempfindlichkeit, sodass Betroffene häufig selbst an bewölkten Tagen eine Sonnenbrille tragen müssen.
Wie sich nephropathische Cystinose im Laufe des Lebens zeigen kann, sehen Sie hier:
Wenn Betroffene keine angemessene Therapie erhalten, kommt es bereits im Kindes- oder frühen Jugendalter zu einem endgültigen Verlust der Nierenfunktion. Für eine bestmögliche Prognose ist es wichtig, die Therapie so früh wie möglich zu beginnen und diese lebenslang konsequent durchzuführen.
Juvenile Form
Bei dieser Form der Cystinose haben Patient*innen zumeist ähnliche Symptome wie bei der infantilen Form, diese beginnen jedoch erst im späteren Kindes- oder Jugendalter und sind weniger stark ausgeprägt.
Okuläre Form (adulte Form)
Bei dieser Form lagern sich die Cystinkristalle ausschließlich in den Augen ab, sodass auch die Auswirkungen der Erkrankung auf die Augen beschränkt sind. Die Betroffenen sind sehr lichtempfindlich und tragen, wie auch die von der infantilen und juvenilen Form Betroffenen, auch an bewölkten Tagen eine Sonnenbrille.
- Nesterova G, Gahl WA. Pediatr Nephrol 2013;28:51–59
- Gahl et al. N Engl J Med. 2002;347(2):111–121.
Diagnose
So stellt man Cystinose fest
Wer von nephropatischer Cystinose betroffen ist, zeigt bereits früh erste Symptome. Schon im ersten Lebensjahr fallen betroffene Kinder dadurch auf, dass sie besonders viel und durstig trinken und nicht erwartungsgemäß wachsen und an Gewicht zunehmen. Bei den anderen Formen der Cystinose sind die Symptome weniger auffällig, sodass der Verdacht nicht immer gleich auf der Hand liegt. Insgesamt gibt es drei wichtige Diagnosemethoden, die meistens miteinander kombiniert werden.
Blutuntersuchung
Bei einem der wichtigsten Diagnoseverfahren wird die Konzentration von Cystin in den weißen Blutzellen, den sogenannten Leukozyten, gemessen. Der Cystingehalt gibt auch Aufschluss darüber, ob die Behandlung anschlägt und richtig dosiert ist.
Um die Blutzellen zu untersuchen, gibt es verschiedene Methoden. Die Analysen werden in Deutschland in zwei Speziallaboren durchgeführt (Stoffwechsellabor der Medizinischen Hochschule Hannover und Stoffwechsellabor der Universitäts-Kinderklinik Münster).
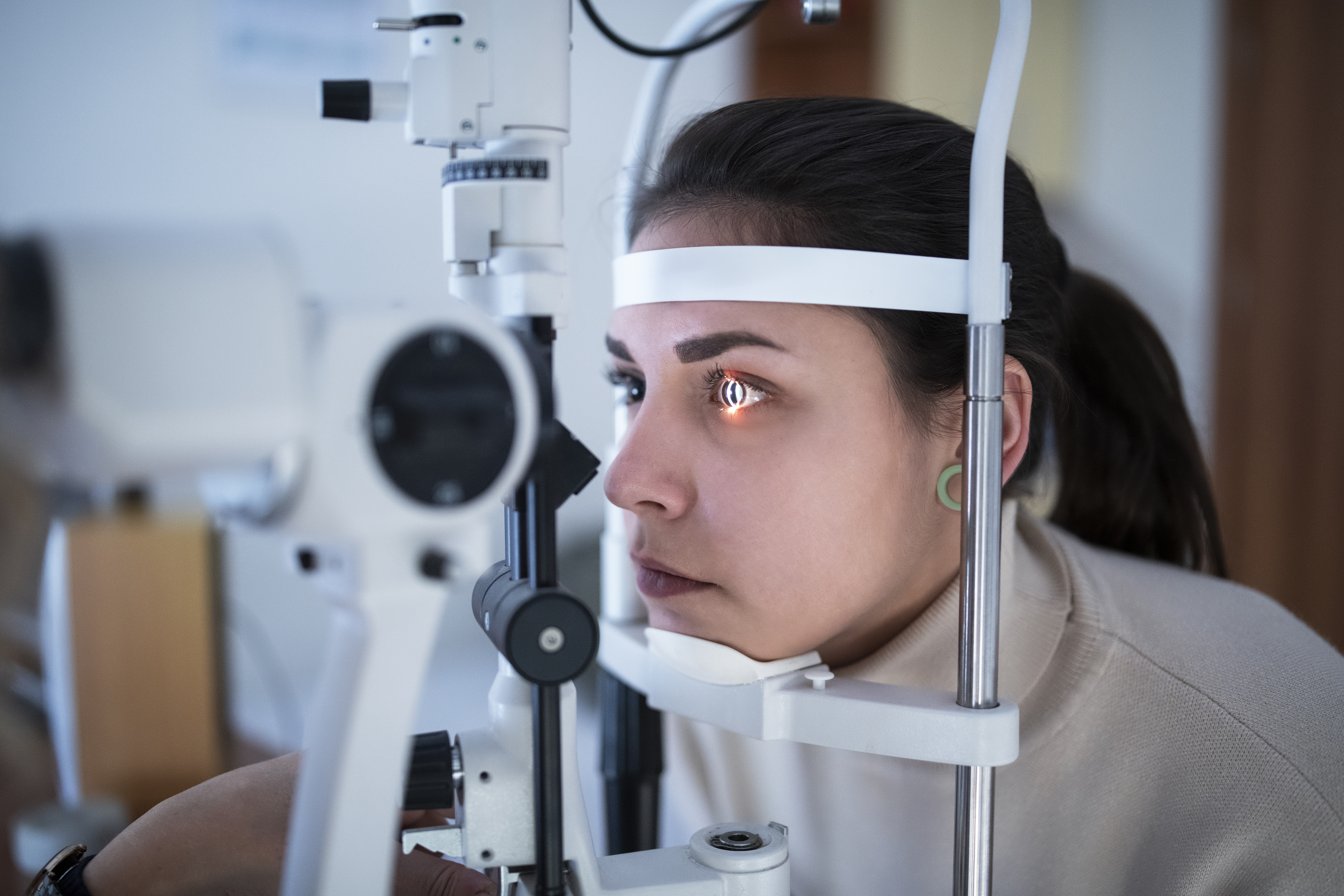
Spaltlampenuntersuchung
Unabhängig vom Alter des*der Betroffenen werden mithilfe einer Spaltlampe Ablagerungen von Cystinkristallen in der Hornhaut sichtbar gemacht, die für alle Formen der Cystinose ab einem Alter von ca. 1 bis 2 Jahren typisch sind.1,2 Die Ablagerungen in der Hornhaut sind außerdem das erste Anzeichen für die bei Erwachsenen auftretende Erkrankung, die sogenannte adulte Form, die in der Regel nur die Augen betrifft.
Genetischer Test
Durch die Blut- und Spaltlampenuntersuchung ist die Diagnose eigentlich schon klar gestellt. Die endgültige Bestätigung erhält man allerdings, wenn die Ursache der Erkrankung, also die zugrunde liegende Mutation, über einen genetischen Test bestimmt wird. Dieser Test dient also zum einen der Bestätigung der Diagnose, zum anderen um die Mutation näher zu bestimmen, die der Cystinose zugrunde liegt.
- Wilmer MJ, et al. Pediatr Nephrol. 2011;26(2):205–215.
- Nesterova G, Gahl W. Pediatr Nephrol. 2013;28(1):51–59.
Verlauf und Prognose
So verläuft Cystinose
Cystinose ist äußerst vielfältig und keinesfalls eine reine Nierenerkrankung, auch wenn die Niere bei der nephropathischen Form besonders früh betroffen ist.1 In der Regel machen sich zwischen dem 6. und dem 12. Lebensmonat allgemeine Beschwerden bemerkbar wie Fieber, Erbrechen, Appetitlosigkeit, Gewichtsstillstand und chronische Verstopfung.1,2 Zudem kommt es zu erhöhter Urinausscheidung (Polyurie), krankhaftem Durst (Polydipsie), Fehlwachstum und einer Rachitis. Die betroffenen Babys und Kleinkinder leiden zusätzlich unter Kraftlosigkeit und erheblichen Entgleisungen des Stoffwechsels, besonders wenn Infektionen hinzukommen. Im Verlauf der Krankheit beschleunigt sich das Geschehen. Zwischen dem 5. und 10. Lebensjahr kann es zu einer Schilddrüsenunterfunktion kommen, während zwischen dem 8. und dem 12. Lebensjahr rasch Nierenschäden auftreten und Lichtempfindlichkeit einsetzt.2,3 Zusätzlich können ab dem 10. Lebensjahr bis zum 30. Lebensjahr Magen-/Darmbeschwerden in Erscheinung treten.2 Dank der Cystin-entspeichernden Therapie und Fortschritte in der Transplantationsmedizin ist die Lebenserwartungen inzwischen deutlich gestiegen.1 Nach einer Nierentransplantation bilden sich in den Zellen des transplantierten Organs keine Cystin-Kristalle. Es konnten jedoch Kristalle nachgewiesen werden, die auf eingewanderte Zellen der Empfänger*innen zurückzuführen sind. Unabhängig von einer Transplantation können insbesondere ohne entsprechende Behandlung, zunehmend Komplikationen auftreten. Dazu gehören beispielsweise erhöhter Blutdruck im Lungenkreislauf, Diabetes, Muskelschwund, Erblindung oder Auswirkungen auf das zentrale Nervensystem.1–3
Die Prognose ist abhängig von der Form der Erkrankung, wobei die nephropathische Cystinose (infantile Form) den schwersten Verlauf zeigt.4 Die Prognose hat sich in den letzten Jahrzehnten deutlich verbessert. Ein früher Therapiebeginn ist entscheidend für den Krankheitsverlauf.5 In einer Studie zeigt sich, dass Patienten*innen, bei denen die Therapie bereits vor dem 5. Lebensjahr eingeleitet wurde, eine höhere Lebenserwartung hatten. 5 Mehr zu der Therapie der Cystinose erfahren Sie hier.
- Nesterova G, Gahl WA. Pediatr Nephrol 2013;28:51–59.
- Gahl WA et al. N Engl J Med. 2002;347(2):111–21.
- Gahl WA et al. Ann Intern Med. 2007;147:242–250.
- Elmonem et al. Orphanet J Rare. 2016;11:47.
- Brodin-Sartorius A et al. Kidney Int. 2012;81(2):179–189.
Therapie und Nebenwirkungen
Behandlungsmöglichkeiten bei Cystinose
Cystinosepatienten*innen werden unter verschiedenen Gesichtspunkten behandelt: Die übergeordnete Cystin-entspeichernde Therapie sorgt dafür, dass die Cystin-Level möglichst niedrig gehalten werden, um das Fortschreiten der Erkrankung zu verlangsamen. Ergänzend wird mithilfe der Therapie des Fanconi-Syndroms der Wasser- und Elektrolythaushalt im Gleichgewicht gehalten. Sie kann auch eine Dialyse oder Nierentransplantation umfassen. Zusätzlich kommt die Behandlung von Symptomen zum Einsatz, die nicht die Niere betreffen, z. B. Wachstumsstörungen.
Cystin-entspeichernde Therapie | Unterstützende Therapie des Fanconi-Syndroms | Behandlung von Symptomen, die nicht die Niere betreffen |
|
Dazu gehört unter anderem:
| Dazu können gehören:
|
Die Cystin-entspeichernde Therapie
In der Cystin-entspeichernden Therapie setzt man auf den Wirkstoff Cysteamin, mit dessen Hilfe das in den Lysosomen gespeicherte Cystin entfernt werden kann. Lesen Sie hier nach, was bei Cystinose im Körper passiert.
Cysteamin gelangt über einen Transporter in die Zellorganellen. Hier sorgt es dafür, dass die schädlichen Kristalle geteilt und über einen anderen Zellausgang abtransportiert werden, um weiteren Schaden zu verhindern. Orales Cysteamin kann die Cystin-Level in Leukozyten auf diese Weise um bis zu 95 % senken.1&
Auf diese Weise kann das Fortschreiten der Erkrankung verlangsamt werden, indem beispielsweise die Funktion der Nieren und anderer Organe länger erhalten bleibt. Eine Cysteamintherapie muss von den Patienten*innen ein Leben lang regelmäßig eingenommen werden, auch wenn sie eine Nierentransplantation erhalten sollten.2,3
Cysteamin ist in zwei verschiedenen Formen als Kapseln verfügbar und sollte entweder zwei- oder viermal täglich eingenommen werden. Zusätzlich wird es als Augentropfen angewendet, um auch die Cystinkristalle in den Augen aufzulösen, die durch das orale Cysteamin nicht erreicht werden. Zu Beginn sollten die speziellen Augentropfen viermal am Tag angewendet werden. In der Regel verbessern sich nach ein paar Wochen die Lichtempfindlichkeit und Augenbeschwerden.
Je früher mit der Cystin-entspeichernden Therapie begonnen wird, desto länger kann die Nierenfunktion erhalten werden und desto besser ist die Lebenserwartung der Patienten*innen. Dafür ist es wichtig, die Cystin-Level durch die regelmäßige Einnahme der Medikamente niedrig zu halten.
Mögliche Nebenwirkungen
Cysteamin ist eine schwefelhaltige Verbindung, wodurch es einen unangenehmen Geschmack und Mund- sowie Körpergeruch verursachen kann. Während der Behandlung kann es auch zu Magenproblemen, Erbrechen oder Durchfall kommen. Alle auftretenden Nebenwirkungen sollten mit dem behandelnden Arzt/mit der behandelnden Ärztin besprochen werden. In manchen Fällen müssen zusätzliche Medikamente (z. B. Magensäureblocker) verschrieben werden oder andere Maßnahmen ergriffen werden.
Langzeittherapie und Routineuntersuchungen
Eine Cystinose-Behandlung begleitet Patienten*innen ein Leben lang. Sowohl Kinder als auch Erwachsene gehen regelmäßig zu Routineuntersuchungen. In der folgenden Grafik sehen Sie, welche Untersuchungen in welchen Abständen stattfinden sollten:
Beispiel für Routineuntersuchungen bei Kindern | Beispiel für Routineuntersuchungen bei Erwachsenen |
| (Bitte besprechen Sie die geplanten Untersuchungen mit Ihrem Arzt/Ihrer Ärztin, die Anzahl oder Art der Untersuchungen kann im Einzelfall abweichen.) | |
Viermal im Jahr:
Jährlich:
| Mindestens einmal im Jahr:
|
- Gahl WA et al. Ann Intern Med 2007;147:242–250.
- Elmonem MA et al. Orphanet J Rare Dis 2016;11:47.
- Levtchenko EN, et al. Pediatr Nephrol. 2006;21(1):110–113.
- Gahl WA et al. N Engl J Med. 2002;347(2):111–21.
- Pisoni RL et al. J Biol Chem. 1995;270(3):1179–1184.
- Langman CB et al. Clin J Am Soc Nephrol 2012;7:1112–20 [Erratum in Clin J Am Soc Nephrol. 2013;8:468].
- Jézégou A et al. PNAS. 2012;109(50):E3434-43.